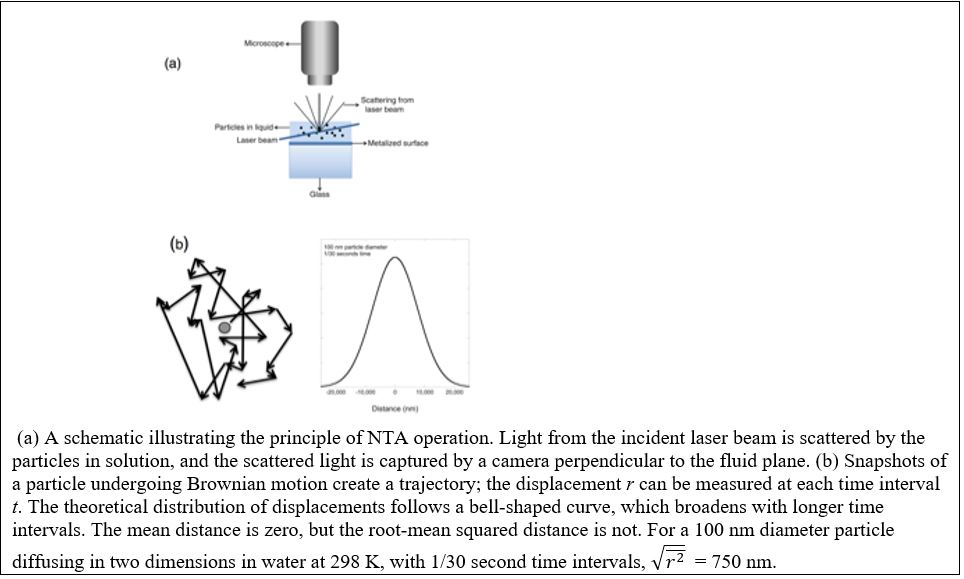
Current Research
Over the last decade, our research program has focused on experimental studies of amyloid protein aggregation, an area of substantial interest because of the role of these protein aggregates in the pathology of Alzheimer’s disease, Parkinson’s disease, and other neurodegenerative disorders.
Transthyretin-mediated inhibition of beta-amyloid aggregation and toxicity.
“Amyloid” is a term describing fibrillar protein aggregates. Although observed in brain tissue of patients with neurodegeneration more than 100 years ago, it was not till the mid-1980’s that the protein associated with Alzheimer’s deposits, beta-amyloid (Aβ), was sequenced, and it was proposed that Aβ aggregation into amyloid fibrils was the underlying cause of the disease. Transgenic mice overexpressing human Aβ were produced in efforts to develop an animal model of Alzheimer’s disease. Despite the presence of significant Aβ deposits, mice did not show typical Alzheimer pathology, raising the question as to the validity of the ‘amyloid hypothesis’. In the mid-2000s, a spontaneous several-fold increase in transthyretin (TTR) was discovered in these transgenic mice, and subsequent investigations showed that the increase in TTR synthesis was responsible for the lack of neuronal pathology. We have successfully demonstrated that TTR sequesters Aβ oligomers and prevents their further aggregation, and that TTR binding to Aβ and sequestration is directly responsible for inhibition of Aβ toxicity. We have identified the specific amino acids on TTR that are required for binding to Aβ. Our group has synthesized novel cyclic conformationally constrained peptides that mimic the Aβ-binding domain on TTR, and we have also discovered that these compounds inhibit Aβ toxicity in vitro, are chemically stable, and retain better selectivity for Aβ in biological fluids than does TTR.
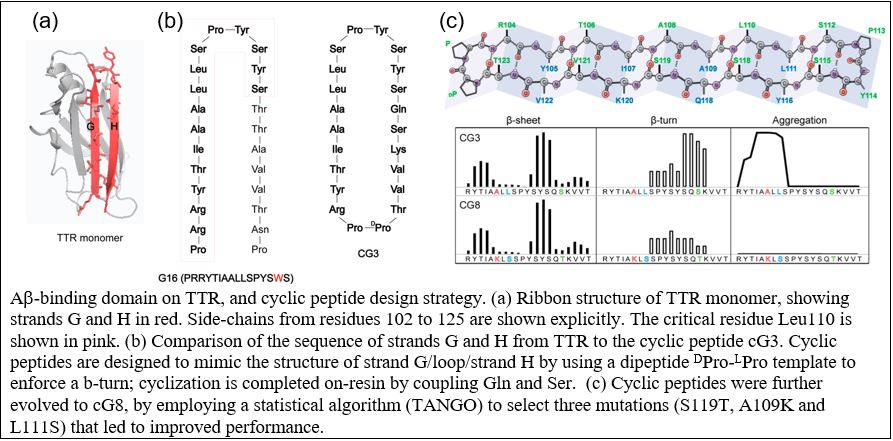
In work currently underway, we are developing strategies for applying these fundamental discoveries to the clinic. First, we are evaluating the method of delivery, tissue uptake, and efficacy of the cyclic peptides in a mouse model. Second, using a distinctly different therapeutic approach, we are testing whether viral-mediated delivery of monomeric TTR to astrocytes can provide a constant source of Aβ-binding TTR. Finally, we are evolving our compounds to achieve higher potency and specificity using computational design and novel hyperstable folded scaffolds.
Beta-amyloid impact on retinol transport across the blood-brain barrier
Retinol (vitamin A) is a critical nutrient for brain health, and there is evidence that retinol supply is limited in Alzheimer’s patients. TTR’s normal function is to transport retinol from the liver (where it is stored postprandially) to target tissues. Two mechanisms have been proposed to explain how retinol enters cells: (1) dissociation of retinol from TTR followed by passive diffusion of the hydrophobic compound across the lipid bilayers, or (2) active transport facilitated by TTR through a channel created by the membrane-embedded protein STRA6.
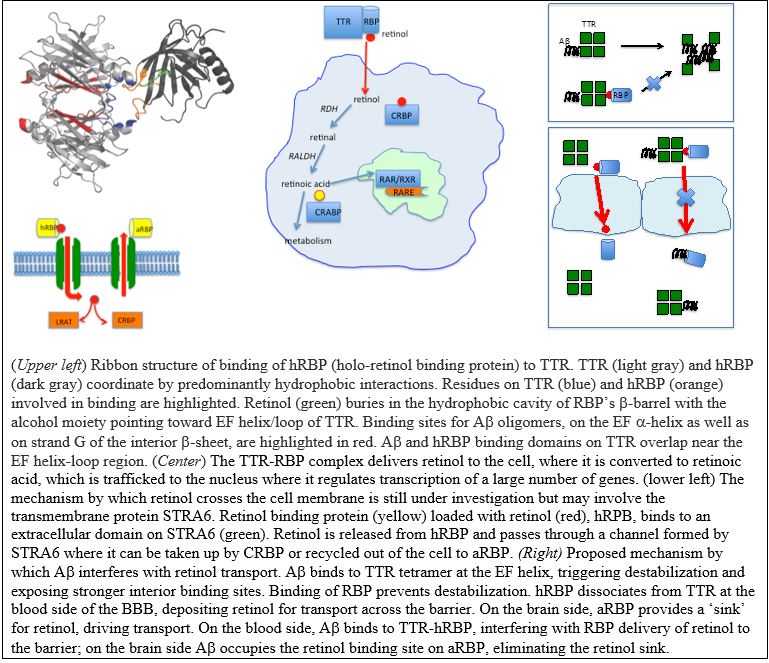
There is virtually nothing known about the mechanism by which TTR facilitates retinol transport across the blood-brain barrier (BBB), but it likely involves one or both of the aforementioned mechanisms – first to transport retinol from the blood into the cells of the BBB, and second to transport retinol out of the BBB cells and into the brain. We have demonstrated that binding of retinol to TTR (via retinol binding protein) competes with Aβ binding to TTR, so we have proposed the novel hypothesis that Aβ binding to TTR may interfere with normal retinol transport across the BBB, which could be a contributing factor to Alzheimer’s pathology. In collaboration with Eric Shusta, we are using a novel in-vitro blood-brain barrier model to quantify the impact of beta-amyloid on retinol transport to the brain.
Cystatin C and the amyloid regulatory network
Several neurodegenerative disorders other than Alzheimer’s have been linked to amyloid protein deposits. These proteins differ in sequence and native structure, but all misfold into aggregates that cause disease. We have been investigating cystatin C (cysC), a cysteine protease inhibitor that is important for maintenance of neuronal health and that can deposit as amyloid in cerebral amyloid angiopathy. CysC inhibits the protease cathepsin B (catB), thus preventing excessive protein degradation; cysC inhibition of catB is believed to play a role in regulating neuronal apoptosis, and high levels of catB are associated with neurodegenerative disorders.
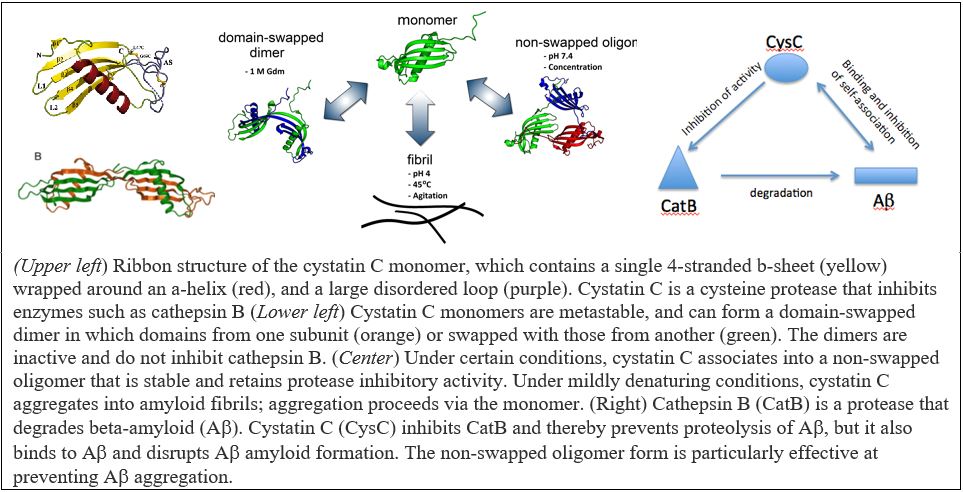
We discovered that cysC can self-associate into soluble oligomers that have some properties of pre-amyloid intermediates, but are actually off-pathway for full amyloid fibril formation. Surprisingly, cysC oligomers (but not monomers) are potent inhibitors of beta-amyloid (Aβ) aggregation. In other words, there may be a regulatory network that controls amyloidogenesis via heterotypic (non-self) interactions. Furthermore, catB degrades Aβ into non-fibril forming fragments, but cysC inhibits this catB activity. Adding to the complexity of this three-protein system, cysC can alternatively associate into domain-swapped dimers that are inactive against catB. In Aβ, catB, and cysC, we have a fascinating system in which protein folding, misfolding, aggregation, binding, degradation, and inhibition are intertwined. Through a combination of experiment and kinetic modeling, we aim to define these relationships quantitatively, and to delineate the regulatory network of cysC-Aβ-catB interactions. Understanding the nature of these complex relationships may have broad repercussions because of the important role of this system in neurodegenerative disorders, cardiovascular disease, cancer, and immunity.
Mechanism of lipid-mediated catalysis of alpha-synuclein aggregation
Parkinson’s disease is a neurodegenerative disorder that has been linked to aggregation of α-synuclein, a protein that is natively disordered. There has been intense interest in the role of lipids in triggering aggregation of α-synuclein, because in the absence of pre-formed ‘seeds’ or lipids, the protein is not particularly aggregation-prone. In contact with membranes, α-synuclein binds to the lipid bilayer, adopts an α-helical conformation and, under certain circumstances, will readily associate into amyloid fibrils. Recent investigations have suggested that aggregation is faster when the lipid bilayer is composed of phospholipids with shorter hydrocarbon chains. However, whether this is a function of the solubility of the phospholipids or the geometry of the bilayer remains an unanswered question. We are exploring the mechanism by which lipid bilayers induce α-synuclein aggregation and the role of chain length and oxidation on aggregation kinetics. As part of this effort, we are developing new methodologies to use a single-particle technique called nanoparticle tracking analysis, (NTA), to quantify aggregation kinetics.